

Fique atento a esta causa de anemia: Mielofibrose – Sabe aquele quadro de anemia que você pode estar tratando há algum tempo e não melhora? Principalmente se esse quadro vier acompanhado de aumento na contagem de plaquetas, pode ser um caso de Mielofibrose Primária ou Idiopática (MF).
A Mielofibrose Primária é um tipo de Neoplasia Mieloproliferativa Crônica (NMPC), grupo de doenças com características semelhantes e que já discutimos em outro post. (clique aqui para ler mais)
Estima-se que a MF tenha uma incidência de 0,5 a 1,5 casos por 100.000 habitantes por ano, ou seja, é uma doença relativamente rara. Acomete pessoas mais idosas, em geral, acima dos 60 anos. Aproximadamente 5 e 17% dos pacientes são diagnosticados antes dos 40 e 50 anos, respectivamente.
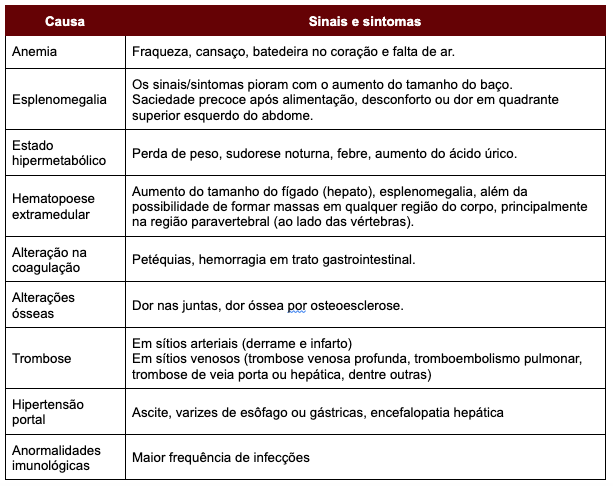
Um quarto dos pacientes não apresenta nenhum sintoma e são identificados quando o hemograma é solicitado como avaliação de rotina. Quando existe alteração, a mais comum é o aumento do baço ou esplenomegalia (identificado pelo exame físico). Os demais casos, os que apresentam sintomas, em geral são secundários à:
Tabela 1: Sinais e sintomas associados ao quadro de Mielofibrose Primária.
É uma doença da medula óssea, também conhecida como o tutano do osso. A medula óssea é composta de medula vermelha (que é onde a produção do sangue acontece) e medula amarela (que é formado por células de gordura).
Figura 1: Imagem representativa da medula óssea vermelha e amarela.

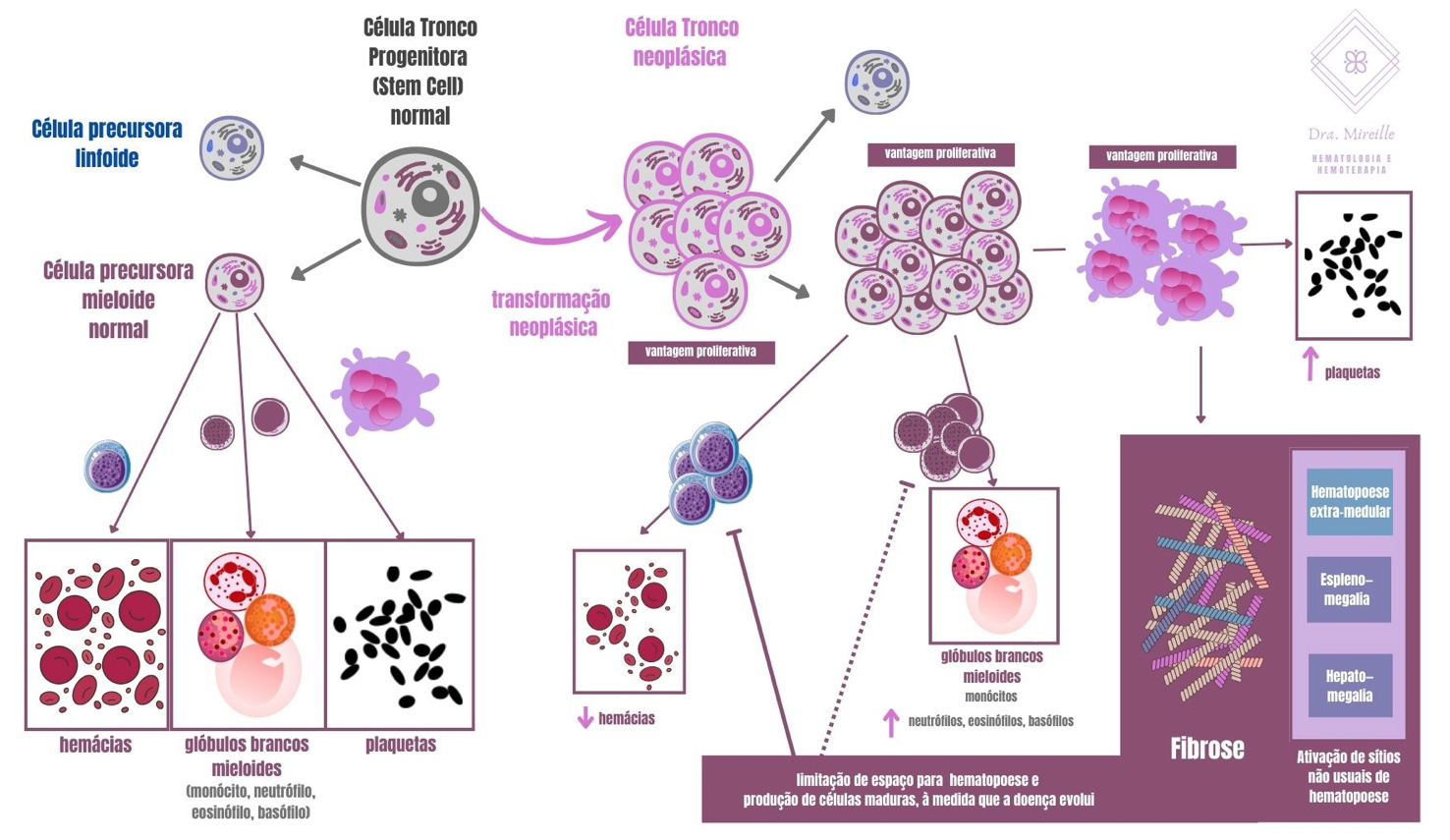
A doença acontece na medula vermelha e tem duas fases: fase pré-fibrótica, inicial, onde a medula óssea se encontra hipercelular, o que evolui até a quase substituição do tecido da medula óssea por fibras (fase fibrótica). Nesta última fase, é como se uma grande cicatriz tomasse todo o espaço de formação do sangue na medula óssea, que tem sua produção comprometida. Com isso, o corpo reativa antigos locais de hematopoese (formação do sangue) fetal, como fígado e baço, além de outras regiões, na tentativa de manter a produção de sangue, esse fenômeno é o que chamamos de hematopoese extramedular.
Na MF, a fibrose ocorre por proliferação clonal da célula tronco, onde todas as células são idênticas à que sofreu o evento neoplásico, o que dá vantagem proliferativa a estas células, mantendo a capacidade de se diferenciar nas formas maduras (ao contrário das leucemias aguda, por exemplo, onde a célula neoplásica perde a capacidade de se diferenciar). Esta alteração genética e adquirida leva ao aumento dos megacariócitos (células precursoras das plaquetas), que liberam fator de crescimento fibrogênico. Tal evento é visto na medula óssea (material coletado por biópsia), como mieloproliferação crônica e hiperplasia megacariocítica atípica. A fibrose da medula óssea é a marca registrada da MF e contribui para a hematopoiese prejudicada que leva à anemia grave.
Figura 2: Esquema representativo dos eventos que levam à mielofibrose.
A mutação JAK2 V617F é um marcador genético, que tem sido detectada em cerca de 50% dos pacientes, mas este marcador não é exclusivo de MF e está presente em outras neoplasias mieloproliferativas crônicas. Além disso, a mutação no receptor do fator de crescimento trombopoetina (MPL) foi observada em 9% dos pacientes e mutações em um gene denominado CARL foi detectado em 20-25% dos pacientes. Esses achados, muito mais que o interruptor que desencadeia a doença, são consideradas alterações que acompanham a doença. Ou seja, muito ainda precisa ser compreendido.
Quando nenhum destes genes é encontrado, a definição da doença se torna mais complexa e é necessário afastar outras situações que cursam com fibrose da medula óssea. O primeiro passo é diferenciar a forma aguda da forma crônica. Para isto, solicitamos hemogramas anteriores e verificamos se a alteração já estava presente anteriormente. A MF é uma doença crônica de instalação lenta (anos). Se o quadro se instalou rapidamente é importante avaliar a possibilidade de mielofibrose aguda, que é uma forma rara de leucemia mieloide aguda (LMA). Nos casos onde a instalação lenta é confirmada, é importante afastar a possibilidade de neoplasia mieloproliferativa Philadelphia (Ph) positivo (leucemia mieloide crônica), o que é feito por estudo citogenético/molecular. A presença de diseritropoese (displasia do setor eritroide) com ou sem monocitose chama atenção para um grupo de doenças chamado de neoplasia mieloproliferativa/mielodisplasia (NMP/SMD). Outras neoplasias mieloproliferativas crônicas Ph negativo, como a polictemia vera (PV), trombocitemia essencial (TE) devem ser consideradas. É necessário ficar atento, também, a outras condições hematológicas, como leucemia de células pilosas (tricoleucemia), linfoma e mieloma múltiplo também devem ser avaliados. Além disso, neoplasias malignas metastáticas para a medula óssea, doenças autoimunes (como LES, esclerodermia, polimiosite e outras), hipertensão pulmonar primária, hiperparatireoidismo secundário e osteodistrofia renal precisam ser avaliados como diagnóstico diferencial.
Com relação ao tratamento, a maioria dos pacientes não vai precisar dele por muitos anos. Alguns, com anemia mais grave, precisarão receber transfusão de sangue. Outros, receberão um quimioterápico oral para controle do aumento de células no sangue periférico (detectado no hemograma). Alguns poucos, terão doença mais agressiva, que é medida por um conjunto complexo de critérios pré-estabelecidos, e poderão receber um medicamento que bloqueia a sinalização na via da JAK, que, como já falamos antes, está implicada no processo da doença.
Para pacientes com doença de alto risco, principalmente os mais jovens e que tenham doador, o transplante de células tronco deve ser considerado como a melhor opção de tratamento. A elegibilidade do transplante varia entre as instituições e geralmente é baseada na aptidão médica e no desempenho funcional. Não existe uma idade específica acima da qual o risco supere claramente os benefícios em pacientes com MF de alto risco.
Em geral, utiliza-se, para todos os casos, um antiagregante plaquetário com o intuito de evitar as tromboses, que acabam sendo as complicações mais temidas. Alguns pacientes que já tiveram trombose venosa, podem necessitar de anticoagulação.
Fique atento a esta causa de anemia: Mielofibrose
Texto elaborado e revisado por
Dra. Mireille Guimarães Vaz de Campos
Médica do corpo clínico do INGOH
Especialista em Hematologia – Hemoterapia
CRM-GO 12.406/RQE 22965.
Texto revisado em Junho de 2022.
É permitida a reprodução parcial
ou total desta obra, desde que
citada a fonte e que não seja para
venda ou qualquer fim comercial.
Referências:
-
CHAUFFAILLE MLLF. Neoplasias mieloproliferativas: revisão dos critérios diagnósticos e dos aspectos clínicos. Rev. Bras. Hematol. Hemoter. 2010;32(4):308-316 (Principal, mas os critérios foram atualizados na última versão do livro da OMS)
-
FIGUEIREDO MS, KERBAUY J, LOURENÇO DM. Hematologia – Guias de Medicina Ambulatorial e Hospitalar UNIFESP-EPM. Manole, 2011.
-
ZAGO MA, FALCAO RP, PASQUINI R. Tratado de Hematologia. Atheneu, 2014.
-
SWERDLOW SH et al. WHO classification of Tumors of Haematopoietic and Lymphoid Tissue: IARC: Lyon 2017
-
TEFFERI A. Management of primary myelofibrosis (2022). In: LARSON RA, ROSMARIN AG. UpToDate. Disponível em: https://www.uptodate.com/contents/management-of-primary-myelofibrosis?search=myelofibrosis&topicRef=4526&source=see_link#H578018087 Visitado em junho de 2022. Fique atento a esta causa de anemia: Mielofibrose


