

Linfoma do Manto: saiba mais sobre esse tipo raro de linfoma não-Hodgkin de células B. – Quem está mais atento ao assunto linfoma sabe que existem mais de 50 subtipos e cada um deles se comporta de forma peculiar. Neste texto iremos abordar os Linfomas do Manto, mas se quiser ler sobre linfomas de uma forma mais geral, clique aqui 1, e se quiser entender melhor a classificação de linfomas e leucemias linfoides, clique aqui 2.
Como surge o linfoma do manto
Os linfócitos são soldados de defesa do organismo que nascem na medula óssea (tutano no osso), circulam pelo sangue e estão predominantemente armazenados nos gânglios linfáticos (linfonodos). Dentro do linfonodo, os linfócitos se colocam em posições específicas de acordo com sua função. Assim, microscopicamente, os linfócitos B se dispõem dentro de folículos e os linfócitos T se dispõem ao redor destes folículos, todos dentro da zona cortical dos linfonodos.
Dentro dos folículos, os linfócitos B entram em contato com o antígeno, geralmente proveniente de um microorganismo e que é o estimulante da resposta imune. Os linfócitos B são ativados e formam uma região chamada de centro germinativo. Ao redor do centro germinativo, mas ainda dentro do folículos (ou seja, ainda em uma zona de linfócitos B) está a zona do manto. Esta zona é formada por aqueles linfócitos B que ainda não tiveram contato com o antígeno estimulador da resposta imune, chamadas de naive (virgens), porque ainda não encontraram antígeno. A maioria dos casos de Linfoma de células do Manto (LCM) são derivados de células B pré-centro germinativo naive (virgens) da zona do manto, enquanto um subconjunto pode se originar da zona marginal ou células B de memória do sangue periférico.
Figura 1: Estrutura esquemática de um linfonodo.

Quando uma pessoa tem linfoma, a célula maligna linfoide começa a produzir cópias idênticas de si mesma (clones) e frequentemente causam o aumento dos linfonodos. Também podem apresentar massas em outras partes do organismo, já que o tecido linfoide está presente em todo o nosso corpo. Há mais de um século, os linfomas foram classificados como tendo as células de Reed-Sternberg (RS), ao que foi chamado de Doença de Hodgkin, ou aqueles que não tinham a célula de RS, chamados de Linfomas Não-Hodgkin. O Linfoma do Manto é considerado um tipo de Linfoma Não Hodgkin.
Epidemiologia
O Linfoma de células do manto (ou Linfoma do Manto, LCM) representa cerca de 7% de todos os linfomas não Hodgkin de células B, ou seja, é muito menos frequente que os outros linfomas, como o Linfoma Difuso de Células B e o Linfoma Folicular. Embora seja frequentemente discutido em conjunto com as formas indolentes de LNH, seu comportamento é mais frequentemente o de um linfoma agressivo. Geralmente compromete mais homens idosos e a idade mediana ao diagnóstico é de 68 anos.
Quadro Clínico (quais os sinais da doença)
A maioria das pessoas (75%) com Linfoma do Manto nota, à princípio, um ou mais linfonodos aumentados de tamanho. Esses linfonodos inchados podem estar em qualquer parte do corpo, como pescoço, virilha ou dentro da barriga. Também podem apresentar febre, perda de peso, suores noturnos que encharcam as roupas, dor, além de cansaço e fraqueza. Além disso, cerca de 25% dos casos se apresentam com doença extranodal (fora dos linfonodos), sendo a maioria dos casos no trato gastrointestinal.
Do ponto de vista molecular
Está associado a alterações nos mecanismos naturais de reparo de dano de DNA, principalmente comprometendo a proteína chamada ciclina D1, envolvida nos processos de controle da apoptose. A apoptose é um mecanismo de morte programada, que todas as nossas células possuem, e que ativa proteínas específicas caso algum problema seja identificado na divisão celular (mutações, quebras de DNA ou outros erros de replicação). A ciclina D1 não é expressa em linfócitos B normais. A expressão da ciclina D1 promove a transição do ciclo celular da fase G1 para a fase S e proliferação, mas a expressão da ciclina D1 por si só não é suficiente para a transformação de linfócitos B normais. Além da expressão da ciclina D1, os tumores LCM demonstram uma diminuição da resposta ao dano ao DNA e aumento da sobrevida celular (apoptose prejudicada). Alterações nessas três vias (ou seja, ciclo celular, reparo de DNA e apoptose) são, portanto, importantes para a transformação neoplásica.
Figura 2. Modelo hipotético dos principais subtipos de linfoma de células do manto (LCM).
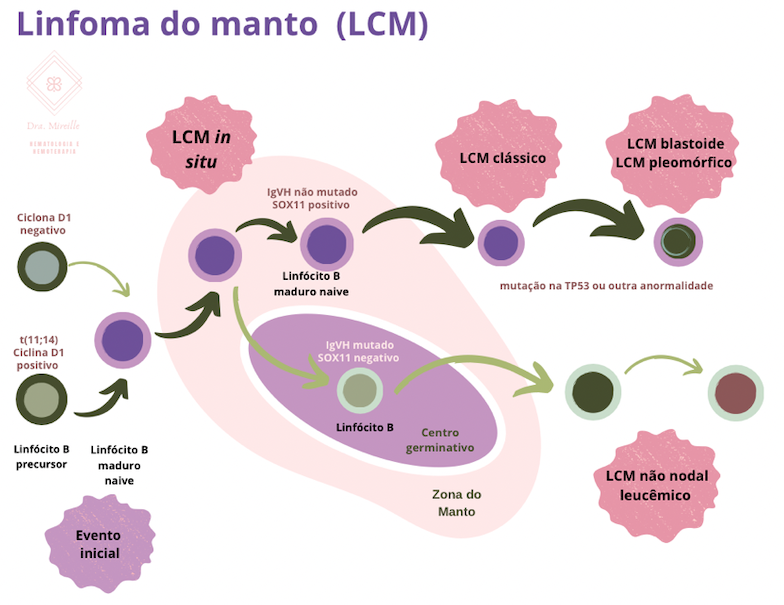
Postula-se que a maioria dos casos de LCM derive de células B pré-centro germinativo naives (virgens) da zona do manto, enquanto um subconjunto de LCM pode se originar da zona marginal ou células B da memória do sangue periférico. A ancestralidade pré-centro germinativo da maioria dos LCMs é apoiada principalmente pela ausência de mutações somáticas nos genes da região variável de cadeia pesada da imunoglobulina (IgVH), que servem como um marcador de trânsito pelo centro germinativo. As células B podem colonizar a porção interna da zona do manto, representando a neoplasia de células do manto in situ (LCMis). Após a aquisição de anormalidades genéticas e moleculares adicionais, o LCMis pode progredir, envolvendo ou não o processo de trânsito pelo centro germinativo (CG), para a forma clássica ou para a forma não nodal leucêmica, respectivamente. Mais frequentemente, o LCM clássico, mas também o LCM não nodal leucêmico podem sofrer anormalidades moleculares/citogenéticas adicionais que levam à progressão clínica e, às vezes, morfológica. Adaptado de Swerdlow et al. e Roue et al.
O LCM está associado a uma alteração citogenética clássica e adquirida (não é herdada dos pais), que é a translocação recíproca entre um pedaço do cromossomo 11 e do cromossomo 14. Esta translocação coloca um gene muito ativado em linfócitos (porção variável do gene da imunoglobulina) junto a um oncogene, que por fim leva à superexpressão da ciclina D1. A demonstração dessa proteína é muito importante na definição diagnóstica do linfoma do manto, já que este pode se “parecer” morfologicamente com outras neoplasias linfoides e a ciclina D1 está positiva em mais de 90% dos casos. As principais neoplasias que podem se parecer com o linfoma do manto são a leucemia linfocítica crônica, o linfoma folicular, o linfoma da zona marginal e o linfoma linfoblástico. Uma minoria dos casos não demonstra ciclina D1, mas muitas vezes super expressa outros mediadores do ciclo celular, particularmente ciclina D2 e ciclina D3. O LCM, incluindo casos negativos de ciclina D1, está frequentemente associado à expressão alterada de SOX11, um fator de transcrição que pode contribuir para um bloqueio na diferenciação em células deste linfoma. A Mutação do TP53, mais do que a deleção do TP53, está associada a um pior prognóstico.
Diagnóstico
Para confirmar o diagnóstico serão necessárias biópsias: do linfonodo, de outro órgão comprometido e de medula óssea. Essas biópsias necessitam ser avaliadas por um patologista especialista em neoplasias hematológicas. Nestes casos, é muito importante que toda a equipe (cirurgião que faz a biópsia, endoscopista, patologista e hematologista) estejam em sintonia e conversem entre si para que este possível diagnóstico não seja negligenciado. O padrão histológico pode ser difuso, nodular ou zona do manto. Alguns casos são descritos como uma combinação dos três padrões. A maioria dos casos é composta exclusivamente de células linfoides de pequeno a médio porte, com núcleos ligeiramente irregulares ou “entalhados”.
O exame de imunohistoquímica, que analisa, além de outros marcadores, a presença da ciclina D1, ajuda muito nesses casos. Alguns dados de imunofenotipagem também ajudam a estabelecer o diagnóstico de linfoma do manto, que é considerado uma doença linfoproliferativa CD5 +. As células do linfoma do manto expressam altos níveis de IgM e IgD de superfície, em geral com expressão da cadeia leve lambda, que é vista em até 80% dos casos. Eles também expressam antígenos de células pan-B (por exemplo, CD19, CD20), CD5 e FMC7. Casos raros podem ser CD5– ou CD23+.
A maioria dos casos irá apresentar a t(11;14), identificada por por citogenética convencional ou hibridização in situ por fluorescência (FISH), embora esse rearranjo não seja específico para LCM. Quando o linfoma do manto envolve o trato gastrointestinal (polipose linfomatosa), as células tumorais expressam a integrina da molécula de adesão alfa-4/beta-7 (CD49d), que normalmente está envolvida no direcionamento de linfócitos para vênulas nos tecidos linfoides associados ao intestino.
Dessa forma, podemos dizer que, em geral, para diagnóstico do LCM se identifica um padrão monomórfico de linfócitos B de pequeno a médio porte com núcleos irregulares e a imunohistoquímica é positiva para translocação de ciclina D1 (BCL1). Alguns casos irão necessitar da citogenética convencional ou FISH demonstrando a t(11;14)(q13;q32) entre o CCND1 e a cadeia pesada de imunoglobulina (IgH).
Estadiamento
Além de uma história clínica e exame físico muito bem executados, a avaliação pré-tratamento inclui estudos laboratoriais, que incluem hemograma completo, bioquímica do sangue com análise de função hepática e renal, além de eletrólitos, desidrogenase lática (DHL), ácido úrico e sorologias para hepatite B e HIV. PET/CT e biópsia de medula óssea também são necessários. Podem ser solicitados outros estudos para sintomas específicos. Dessa forma, punção lombar com análise de liquor (que é o líquido que banha o cérebro) pode ser necessário nos casos com variante blastoide ou quando há sinais de comprometimento do SNC. Endoscopia digestiva alta e colonoscopia são realizadas em pacientes com sintomas gastrointestinais ou achados em exame físico sugestivos de envolvimento gastrointestinal, mas podem ser indicados em assintomáticos quando este envolvimento muda o manejo (por exemplo, doença estágio I/II).
Esses exames servem para estabelecer o quanto o linfoma está comprometendo o corpo e compõem o que chamamos de estadiamento. Usualmente, o LCM é identificado em estadio avançado (estadio IV), envolvendo linfonodos acima e abaixo do diafragma, podendo comprometer as chamadas regiões extranodais (aqui se destaca o comprometimento do trato gastrointestinal) e medula óssea. Além disso, as células doentes podem ser encontradas no sangue periférico, situação que comumente é chamada de linfoma leucemizado.
Tratamento
O tratamento dependerá de alguns fatores como:
- sintomas apresentados,
- localização dos linfonodos comprometidos pelo linfoma,
- idade
- outras condições médicas (doenças cardíacas, renais, diabetes, dentre outras).
Quase todos os pacientes precisam de um estudo inicial da fração de ejeção cardíaca (por exemplo, por ecocardiograma) antes de receber um dos componentes do tratamento, chamado de antraciclina. Antes do início do tratamento, homens e mulheres com potencial para gerar filhos devem receber aconselhamento sobre o eventual efeito do tratamento em sua fertilidade e devem ser aconselhados a considerar medidas de preservação da fertilidade.
A terapia pode incluir:
- quimioterapia, medicamentos que matam as células cancerosas ou impedem que elas cresçam através de ações no ciclo celular;
- radioterapia;
- anticorpos monoclonais, que são proteínas criadas em laboratório e que matam as células cancerígenas usando moléculas de superfície celular como alvo;
- terapia de alvo com inibidores de tirosina quinase visando moléculas expressas dentro da célula B que, em geral, tem ação sobre a tirosina quinase de Bruton que faz parte da via de sinalização do receptor do linfócito B e encontra-se altamente ativada em neoplasias linfoproliferativas.
- cirurgia, que geralmente não é benéfica, mas pode ser valiosa em pacientes que apresentam obstrução intestinal, por exemplo.
Existe um pequeno subgrupo de pacientes que terá um curso mais indolente e pode não necessitar de tratamento inicialmente. No entanto, a maioria dos pacientes necessita de tratamento ao diagnóstico. Em pacientes jovens com boa condição de saúde, pode-se utilizar tratamento com imunoquimioterapia mais agressiva, seguido de transplante de medula óssea autólogo. É importante entender que há grande diversidade de práticas clínicas e ambiguidades em torno da melhor abordagem de tratamento preferencial para pacientes com linfoma do manto.
Em geral o esquema de indução inclui imunoquimioterapia, com um anticorpo monoclonal anti-CD20 (rituximabe, por exemplo) associado a esquema quimioterápico convencional como CHOP, bendamustina e eventualmente esquema mais agressivo como o HYPERCVAD ou esquema alternando CHOP e DHAP.
A resposta ao tratamento é determinada usando informações coletadas de história clínica, exame físico, testes laboratoriais e tomografia computadorizada. Caso tudo estiver como planejado, o regime de primeira linha é continuado, ou se não houver uma boa resposta, o tratamento de segunda linha pode ser oferecido. Se tudo correr bem com o protocolo de primeira linha, são solicitados exames de sangue para avaliar novamente a resposta ao tratamento ao final dos ciclos planejados. Se houver uma boa resposta, a “manutenção” por 2 a 3 anos com um anticorpo monoclonal anti-CD20 (por exemplo, rituximab) em geral é oferecida. Existem inúmeros medicamentos que podem ser considerados em caso de recaída ou refratariedade da doença e vários estudos clínicos estão sendo conduzidos nesta área.
O curso do LCM é moderadamente agressivo e variável. Vários estudos tentaram determinar os fatores prognósticos para prever quais pacientes terão um curso mais agressivo. Os sistemas de pontuação mais comumente utilizados envolvem fatores como idade, nível de DHL, estado geral de saúde (escala ECOG ou performance status), contagem de leucócitos em sangue periférico (hemograma), dentre outros.
O linfoma de células do manto é uma doença rara que pode ser difícil de diagnosticar e difícil de tratar. Apesar de todos os esforços, a terapia não é curativa. Alguns pacientes irão atingir remissão e outros irão necessitar de retratamento de tempos em tempos. Estes pacientes deveriam ser sempre tratados por um especialista com experiência neste tipo de linfoma, para garantir uma boa qualidade de vida, com poucos sintomas associados ao tratamento. A boa notícia é que mais de uma dezena de terapias estão em estudo e algumas alternativas já se mostraram promissoras em segunda linha.
Se você foi diagnosticado com este tipo de linfoma, fale com seu médico sobre qualquer dúvida ou preocupação que tenha e tente entender suas opções de tratamento. Cada caso tem suas particularidades e só seu médico sabe todos os detalhes. Dessa forma, será possível decidir qual tratamento é o mais adequado para você.
Fique atento que o que foi dito aqui só se aplica ao linfoma do manto! Outros tipos de linfoma têm comportamentos diferentes e necessitam de tratamento específico.
Linfoma do Manto: saiba mais sobre esse tipo raro de linfoma não-Hodgkin de células B.
Siga a INGOH no Instagram!
Texto elaborado e revisado por
Dra. Mireille Guimarães Vaz de Campos
Médica do corpo clínico do INGOH
Especialista em Hematologia – Hemoterapia
CRM-GO 12.406/RQE 22.965.
Texto revisado em Junho de 2022.
É permitida a reprodução parcial
ou total desta obra, desde que
citada a fonte e que não seja para
venda ou qualquer fim comercial
Referências:
-
ALAGGIO R , AMADOR, C., ANAGNOSTOPOULOS, I. ET AL. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Lymphoid Neoplasms. Leukemia 2022; 36, 1720–1748. https://doi.org/10.1038/s41375-022-01620-2.
-
BROWN JR, FREEDMAN AS, ASTER JC. Pathobiology of mantle cell lymphoma In: UpToDate, LISTER A & ROSMARIM A(Ed), Waltham, MA. (Visitado em junho de 2022).
-
CHERSON BD ET AL. Recommendations for Initial Evaluation, Staging, and Response Assessment of Hodgkin and Non-Hodgkin Lymphoma: The Lugano Classification. Clin Oncol 2014; 32:3059-3067.
-
FIGUEIREDO MS, KERBAUY J, LOURENÇO DM. Hematologia – Guias de Medicina Ambulatorial e Hospitalar. UNIFESP-EPM. Manole, 2011.
-
FORONES NM ET AL. Oncologia – Guias de Medicina Ambulatorial e Hospitalar UNIFESP-EPM. Manole, 2005; capítulo 44, 321-336. (Classificação clínica)
-
FREEDMAN AS, ASTER JC. Clinical manifestations, pathologic features, and diagnosis of mantle cell lymphoma. In: UpToDate, LISTER A & ROSMARIM A(Ed), UpToDate, Waltham, MA. (Visitado em junho de 2022).
-
FREEDMAN AS, FRIEDBERG JW.. Initial treatment of mantle cell lymphoma. In: UpToDate, LISTER A & ROSMARIM A(Ed), UpToDate, Waltham, MA. (Visitado em junho de 2022)
-
FREEDMAN AS, FRIEDBERG JW. Treatment of relapsed or refractory mantle cell lymphoma. In: UpToDate, LISTER A & ROSMARIM A(Ed), UpToDate, Waltham, MA. (Visitado em junho de 2022)
-
HOFFBRAND AV & MOSS PAH. Fundamentos em Hematologia. 6. Ed. Porto Alegre: Artmed, 2013.
-
HOFFMAN et al, Hematology, basic principels and practice, 4. Edição
-
LONGO, DJ. Harrison’s Hematology and Oncology, 2ed.
-
ROUE G & SOLA B. Management of Drug Resistance in Mantle Cell Lymphoma. Cancers 2020, 12, 1565; doi:10.3390/cancers12061565
-
SWERDLOW SH et al. WHO classification of Tumors of Haematopoietic and Lymphoid Tissue: IARC: Lyon 2017
-
ZAGO MA, FALCAO RP, PASQUINI R. Tratado de Hematologia. Atheneu, 2014.


